本试剂盒由上海金畔生物科技有限公司与留美学者共同开发并在中国申请了组合发明专利。由于大肠杆菌表达系统遗传背景清楚、易于大规模发酵等优点,尤其是近年来采用分子内伴侣的技术(如Trx、DsbA等)使得许多外源基因能在E.coli里正确折叠,如IL-11、GM-CSF、INF、 Insulin甚至包括结构复杂的tPA等均可获得正确折叠的重组可溶性活性蛋白,使得E.coli高效快速表达系统仍然是分子生物学实验室一个不可缺少的工具。
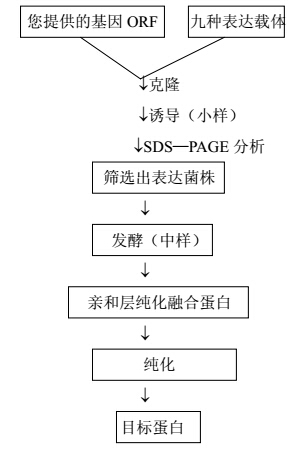
本试剂盒包括冻干菌株、抗生素和统一多克隆位点的九种表达载体。用户只要扩增克隆出一段不带终止密码子或者带终止密码子的阅读框编码基因克隆进九种表达载体,可同时筛选,节省时间。一般两至三周可以筛选出表达菌株,在此基础上,进一步根据需要进行实验室菌株的优化从而成为工程菌株。
5、pET-GST表达载体采用T7启动子和GST融合表达,同时选用人鼻病毒14亚型3C蛋白酶专一性切割位点。本公司有高纯度的重组人鼻病毒14亚型3C蛋白酶出售,同时还有专一性吸附该蛋白酶的树脂出售。此外,C端还添加了6个His,便于采用chelating sepharose亲和层析。本载体表达的融合蛋白可采用吸附GST的亲和层析和6个His的chelating sepharose双重亲和层析,极大地方便了下游纯化。GST融合表达背景文献在medline里极为丰富,有数百个基因采用GST融合表达成功。
6、pET-Trx表达载体采用蛋白质工程改造的硫氧还蛋白即将其中两个氨基酸突变成His,使硫氧还蛋白在空间立体结构上形成三个组氨酸位点(His-patch),便于采用chelating sepharose亲和层析。该表达载体采用T7启动子和(His-patch)Thioredoxin融合表达,同时选用人鼻病毒14亚型3C蛋白酶专一性切割位点。硫氧还蛋白作为分子内伴侣可以帮助许多基因在E.coli胞内正确折叠如IL-3、IL-6、IL-11等。
7、pET-His表达载体采用N端6个His和T7启动子。高效表达产物的N端具备6个His,便于采用chelating sepharose 亲和层析纯化目标蛋白。同时该载体选用了凝血酶专一性切割切点。融合蛋白既可以采用高纯度限制级的冻干凝血酶液体切割,也可以采用固相化凝血酶切割。
8、pTrc-CKS表达载体采用E.coliTrc启动子和蛋白质工程改造过CKS (CTP:CMP-3-deoxy-D-manno-octulosonate cytidylyltransferase or CMP-KDO synthetase)基因融合表达,同时选用人鼻病毒14亚型3C蛋白酶专一性切割位点。CKS融合表达是美国Abbot公司的专利,CKS融合蛋白研制成功HIV和HCV重组抗原并取得FDA认证在美国上市。CKS是一个非常稳定且高度可溶的理想融合伴侣。本发明除去掉CKS C端8个氨基酸外,还将C端Met突变成Thr,一方面可报自已的专利,另一方面有利于基因表达。
9、pET-DsbA表达载体采用T7启动子和DsbA基因融合表达,同时选用凝血酶切割位点。DsbA是近年来发现帮助二硫键形成的重要分子内伴侣之一。本试剂盒DsbA基因去掉了信号肽,同时将DsbA氧化还原功能相关的两个半胱氨酸(cys)突变成丝氨酸(ser),这样与突变后的DsbA融合可实现许多基因的胞内可溶性有活性的高表达(表达量大于30%),如IGF-1、IGFBP、EGF、Insulin等。
二、基因克隆
1、本试剂盒选用两种大肠杆菌菌株
两者基因型分别为:
E.coli TOP10
E.coli BL21(DE3)plysS
注意:E.coli BL21(DE3)plysS 转化菌可以添加氯霉素
2、两种大肠杆菌菌株使用情况
这一点非常重要,务必分清。
(1)九种载体的扩增须采用E.coli TOP10,而不能采用E.coli BL21(DE3)plysS。
(2)九种载体的目的基因克隆须采用E.coli TOP10,而不能采用E.coli BL21(DE3)plysS。
(3)克隆了目的基因的表达载体,其中1、2、3、4和8号载体即pLLP-OmpA、pLLP-STII、pMBP-P、pMBP-C和pTrc-CKS应转化进E.coli TOP10进行诱导表达。5、6、7和9号载体即pET-GST、pET-Trx、pET-His和pET-DsbA应转化进E.coli BL21(DE3)plysS进行表达。
3、原始种子库和实验种子库的建立
一般分子生物学和基因工程实验室常用来扩增质粒的菌株E.coli JM109、E.coli DH5α、E.coli HB101等,常常从别的实验室转种而来,对其基因型缺乏确切的验证。学生在实验研究中常常用LB平板连续传代,有时一个学期就靠平板上传代来培养感受态等试验。这样表面上看来很方便,但事实上由于微生物容易发生突变,这就造成了克隆实验中转化效率不高、转化涂平板时出现氨苄抗性的小菌落、提取的质粒酶切时易于降解甚至出现基因重组突变之类的实验怪现象,这些小问题往往造成实验进程受阻,更糟的是很难分析原因。
为了根本上克服这个问题,我们将建议您购买本试剂盒后第一件事就是建立原始种子库、试验种子库和感受态种子库。
1)E.coli TOP10冻干菌株和E.coli BL21(DE3)plysS冻干菌株,为原始种子库,于-20oC长期保存。
2)从冻干的原始种子库中取一支里面冻干的少许粉末,加入10ml LB液体培养(其中E.coli TOP10F具四环素抗性,10ml LB液体培养基加5ml(浓度为5mg/ml)四环素;E.coli BL21(DE3)plysS为氯霉素抗性,10ml LB液体培养基加5ml(浓度为25mg/ml)氯霉素。)37oC,150转振荡培养过夜。
3)第二天转种1ml至40ml液体LB培养基(抗生素添加方法同上,注意E.coli TOP10F抗四环素,E.coli BL21(DE3)plysS抗氯霉素)。37oC,200转振荡培养3-4小时,至OD600 0.4-0.5之间,取一半用15%的灭菌甘油各保存50支(-70oC),即成试验种子库。另一半用SSCS溶液制备感受态,分装于每支120ml 50-100支于-70oC保存,即成感受态种子库。
4)如没有SSCS溶液,今后可每次从试验种子库里选一支划LB平板,用后即弃去。可用CaCl2法制备新鲜感受态,一般CaCl2法感受态在4oC可保存两至三天。平板上菌株保存时间不超过三周,避免反复连续在LB平板上传代。
4、九种表达载体质粒的扩增
本公司试剂盒里提供的九种表达载体经DNA序列分析、表达试验、酶切分析、热稳定(37oC)等测试,我们建议您克隆目的基因前将上述九种载体转化进E.coli TOP10,自己提质粒,试剂盒的原始质粒于-20oC长期保存。具体实验方法为:
1)制备E.coli TOP10感受态,可采用CaCl2新鲜制备,也可采SSCS溶液制备的-70oC保存感受态。
2)各取1ml九种原始质粒并设负对照一个转化,转化方法参见《分子克隆》。
3)LB固体平板可采用《分子克隆》的配方或营养琼脂均可,注意添加氨苄抗生素。
4)挑单菌落,提取九种载体质粒。
5)根据说明书提供的载体特征大小和酶切位点进行鉴定。
5、目的基因的扩增和酶切位点的选择
1)本试剂盒选用了统一的多克隆位点为
GGA TCC GCA GAA TTC AGC GCT AGC CAC(TAA) CAT CAT
BamHI EcoRI NheI His His His
CAT CAT CAT TAA GCTT
His His His HindIII
我们建议您扩增基因5‘端选BamHI或BglII,3’端选NheI或XbaI或SpeI,同时3‘端不要带终止密码子,注意与GGA TCC(Gly Ser)表达框架正确,载体用NheI-BamHI双酶切。BamHI和BglII为同裂酶,NheI、XbaI和SpeI为同裂酶。
如您目的基因无法满足上述要求,如同时含有BamHI和BglII,5’端可选择EcoRI,但须注意与表达框架GAA TTC(Glu Phe)对齐。如您目的基因同时含有XbaI、NheI和SpeI,3‘端可选择EcoRI。
如果您的目的基因已带有终止密码子,同样可以进行克隆表达,只是注意有些表达载体是在C端带6个His。
PCR引物设计时BamHI和EcoRI位点的5’端保护碱基有2至3个GC即可,而XbaI、NheI和SpeI位点5‘端保护碱基须在5个左右才容易被酶切割。
6、我们建议您PCR产物先克隆至T-载体上,选单菌落进行DNA序列分析正确后,再进行克隆表达。这是由于PCR扩增过程中容易出错,直接进行表达分析相关因素就更复杂了。
7、基因克隆后建议您通过双酶切或PCR或DNA测序鉴定确认。
基因高效快速表达试剂盒载体测序引物
一) pLLp-OmpA
正向测序引物为5 GGC TTT ACA CTT TAT GCT TC 3
反向测序引物为 5CCA TTT TTC ACT TCA CAG G 3
二) pLLp-STII
正向测序引物为5 GGC TTT ACA CTT TAT GCT TC 3
反向测序引物为 5CCA TTT TTC ACT TCA CAG G 3
三) pMBp-P
反向测序引物为通用引物M13/PUC SEQUENCING PRIMER
5 CGC CAG GGT TTT CCC AGT CAC GAC 3
四) pMBp-C
反向测序引物为通用引物M13/PUC SEQUENCING PRIMER
5 CGC CAG GGT TTT CCC AGT CAC GAC 3
五) pET-GST
反向测序引物为通用引物T7 terminator primer
5 GCT AGT TAT TGC TCA GCG G 3
六) pET-Trx
反向测序引物为通用引物T7 terminator primer
5 GCT AGT TAT TGC TCA GCG G 3
七) pET-His
正向测序引物为通用引物T7 promoter primer
5 TAA TAC GAC TCA CTA TAG GG 3
反向测序引物为通用引物T7 terminator primer
5 GCT AGT TAT TGC TCA GCG G 3
八) pTrc-CKS
正向测序引物为5 ATC CAT GTT GCT GTT GCT CAG G 3
反向测序引物为5 GAT TTA ATC TGT ATC AGG 3
九) pET-DsbA
反向测序引物为通用引物T7 terminator primer
5 GCT AGT TAT TGC TCA GCG G 3
三、基因表达
1、载体的选择
您在进行克隆表达前须仔细阅读九种载体的相关文献资料。
第一次实验时,我们建议您先其中四个载体即3号、7号、8号和9号载体进行克隆表达。如果3号载体表达可分泌到周质空间,您可以进行1号和2号载体的直接分泌表达试验。一般8号载体均可以表达且克隆进E.coli Top 10后提取质粒的同时可以进行表达试验。
2、表达培养基(固体)的选择
表达培养基(固体)有两种配方,配方一采用常见的LB配方,这一配方比较常用,大多基因表达均可采用这一配方。注意事项如下:
1)固体平板倒好后一定要在室外温下放置24小时,以使表面变干,否则影响表达。
2)添加0.2%葡萄糖有利于表达。
3)选用试剂的厂家很重要。建议您先用OXOID牌trypton和yeast extract,琼脂Agar采用进口产品。
具体操作如下:
1)称2g trypton、 1g yeast extract、1g NaCl和4g Agar加蒸馏水至198ml,15磅灭菌25分钟。
2)配20%葡萄糖20ml(国产分析纯),8磅灭菌25分钟。
3)待上述两种试剂冷却下来,取2ml 20%葡萄糖加入LB固体培养基中,总计200ml,加氨苄到100-120μg/ml浓度,倒固体培养基约10个平板。
4)上述平板放在室温下放置24小时,以使表面变干。
5)将上述克隆鉴定正确的带目的基因的表达质粒0.5μg-1μg转达化相应受体菌感受态。
6)1号、2号、3号、4号和8号重组质粒转化E.coli TOP 10进行表达,5号、6号、7号和9号重组质粒转化E.coli BL 21(DE3) plysS进行表达。
7)转化热休克→冰浴后添加50μl-100μl soc培养基或LB培养基在摇床35oC 150转培养30分钟涂布。涂布时添加5μl(浓度为1mg/ml)氨苄青霉素一并涂布,有利于表达。
表达培养基(固体)另一种配方为NZ-Amine加无机盐加葡萄糖。这一配方不常用,但能更好地控制诱导前表达的问题。如您遇到对E.coli宿主毒性较大的基因如在普通LB平板上很难生长时,可采用这一配方(注意:一般不需要采用这一配方,LB配方就可以了)。
具体操作如下:
(1)称3.4g NZ-Amine(with MgSO4+NaCl) 加4g进口琼脂加蒸馏水至176ml,15磅灭菌25分钟。即为基因表达培养基成份A。
(2)将2g NH4Cl(无水计)、6g KH2PO4(无水计)和 6g Na2HPO4 (无水计)加蒸馏水至200ml,15磅灭菌25分钟。即为基因表达培养基成份B母液。
(3)配20%葡萄糖20ml,8磅灭菌25分钟。
(4)待上述三种试剂冷却下来,取20ml基因表达培养基成份B母液和4ml 20%葡萄糖,添加至176ml的含2% 琼脂的基因表达培养基成份A中,总计200ml,加氨苄至100-120mg/ml浓度,倒固体培养基约10个平板。
(5)上述平板放在室温下放置24小时,以使表面变干。
(6)将上述克隆鉴定正确的带目的基因的表达质粒0.5-1mg转化受体菌感受态。
(7)1号、2号、3号、4号和8号重组质粒转化E.coli Top 10F’进行表达,5号、6号、7号和9号重组质粒转化E.coli BL 21(DE3)plysS进行表达。
(8)转化热休克→冰浴后添加50μl-10μl SOC培养基或LB培养基在摇床35oC 150转/分培养30分钟后涂布。涂布时添加5μl(浓度为1mg/ml)氨苄青霉素一并涂布,有利于是表达。
3、基因表达测定
液体培养基(LB配方)
每升 tryptone 10g
yeast extract 5g
NaCl 10g
加水至1升,调pH至7.2,15磅灭菌25分钟
配制20%葡萄糖,8磅灭菌20分钟,添加至上述LB中,终浓度为0.2%.
氨苄采用100mg/ml-120mg/ml工作浓度。
诱导剂IPTG母液 100mg/ml
1至3号质粒表达测试方案(周质空间分泌表达)
1)、将上述重组的1至3号质粒转化出单菌落挑至20ml LB培养基(+glucose 0.2% + Amp 120μg/ml,50ml三角瓶加8层纱布)于晚上9:30接种,100转(不要太快),30oC培养(不要37oC),摇至第二天上午8:30。取8ml种子液加至200 ml液体LB(+glucose 0.2% + Amp 120μg/ml,1升三角瓶加8层纱布)35oC 180转快摇至OD600为0.6左右,加IPTG至100μg/ml(IPTG浓度今后可以优化),同时再补充一次新的Amp至120μg/ml(两次共为240μg/ml),30oC诱导3小时。
2)、离心收集菌体,用30mM Tris HCl、20%蔗糖1mM EDTA pH8.0(按每克菌体80ml计)悬浮后冰浴,轻轻振荡10分钟。
3)、8000g离心4oC,去除上清,沉淀用5mM MgSO4 8ml悬浮后冰浴,轻轻振荡10分钟。
4)、12000g离心15分钟,取上清即为cold osmotic shock fluid.
5)、取3种重组质粒和3种原质粒的osmotic shock fluid样品加SDS-PAGE上样缓冲液,走电泳分析。如出现明显的区带且分子量与预计的一致即获得了周质空间分泌表达(MW MBP with linker =45KD)。
4至9号重组质粒基因表达测试(胞内表达)
由于4至9号重组质粒均为胞内表达,且表达量一般均大于20%,所以采用小样20ml LB培养诱导即可将菌体离心走SDS-PAGE 电泳分析。
特别注意,由于本试剂盒5号、6号、7号和9号采用高拷贝数质粒pUC18做母体,其一方面质粒高拷贝数有利于高表达,但另一方面其表达分泌的β-内酰胺酶也很高。而高浓度的β-内酰胺酶水解LB平板上和LB液体里的氨苄。而一旦氨苄消耗掉了,E.coli BL 21(DE3) plysS很容易丢失重组质粒,而不带重组质粒的E.coli 生长得更快从而比例会越来越多,这样发酵过浓后表达会下降甚至很低。为此,您须仔细阅读下面有关转化和发酵的操作要求。(如您需要高密度发酵,建议您在本试剂盒筛选到合适表达方案后将表达元件从启动子至转录终止子扩增克隆到带卡那霉素抗性基因的质粒上,这样可以克服氨苄易被水解的问题)。
具体操作如下:
(1)LB+Amp+glucose固体平板室温下放置24hr后可以使用。
(2)一般上午10:00转化重组质粒5号、6号、7号和9号进E.coli BL21(DE3) plysS(注意:4号和8号尤其是8号一般较稳定,转化平板放2-3天均可以做表达,但5号、6号、7号和9号须转化后立即做表达)。
(3)晚上9:00接种,要求5号、6号、7号和9号转化E.coli BL21(DE3) plysS菌落较小(一般菌落越小,第二天表达量就越高)。4号和8号转化E.coli TOP10F’,接种时对菌落大小要求不高。接种进20ml LB(+glucose 0.2% + Amp 120μg/ml,用50ml三角瓶加8层纱布,纱布通气好,不要用棉花塞子)(特别注意,转速要慢,否则菌体浓度过高,Amp水解了表达会下降)。
(4)70–90转/分钟30oC培养过夜至第二天上午8:30 OD600约为0.5加IPTG 100μg/ml,150–170转/分钟37oC诱导(5号、6号、7号和9号诱导1.5小时,4号和8号诱导3小时)。
(5)离心收集菌体走SDS-PAGE电泳分析。
(6)如出现明显的区带且分子量与预计的一致即获得表达。(MW MBP with linker=45KD, MW GST with linker =28.7KD,MW CKS wih linker=29.5KD,MW TRX with linker=14KD,MW DsbA with linker=24.7KD)。
4至9号重组质粒表达产物可溶性分析:
通过小样(20ml)表达测试后确认的表达菌株采用200ml摇瓶发酵确认表达产是否可溶。具体操作如下:
(1)按上述方法转化、培养20ml过夜种子液(注意严格按照上述方案进行,要注意5号、6号、7号和9号转化E.coli BL 21(DE3) plysS菌落较小时接种,慢摇70–90转/分钟30oC培养过夜)5ml种子液加至200ml LB(+glucose 0.2% + Amp 120μg/ml,用1000ml三角瓶加8层纱布)快摇35oC 180转/分2-3小时至OD600=0.6时加IPTG 100μg/ml诱导(注意采用30oC诱导,5号、6号、7号和9号诱导时间1.5小时即可,4号和8号诱导3小时)。
(2)用PBS约20ml悬浮菌体,超声破碎,适当加少量溶菌酶。
(3)超声液离心12000g,15分钟,上清和沉淀走SDS-PAGE分析(MW MBP with linker=45KD, MW GST with linker =28.7KD,MW CKS wih linker=29.5KD,MW TRX with linker=14KD,MW DsbA with linker=24.7KD)。上清有表达即为可溶部分。沉淀部分既可形成包涵体,也可以就是一般性沉淀。
发酵条件的优化与菌株优化:
1)一般发酵时低温、低浓度诱导剂(IPTG)有利于表达分泌型、可溶型重组蛋白表达,但低温时(一般分为28oC-30oC、23oC-25oC两个区段)诱导时间可延长。
2)对宿主毒性较大的基因表达可加大氨苄的浓度(可达200mg/ml)并提高培养基中葡萄糖浓度(可达0.5%)有利于稳定表达。
3)一般发酵时温度高、诱导剂浓度高有利于表达包涵体型重组蛋白。
4)进一步优化成工程菌株的实验以及中试放大试验,凡购买本公司试剂盒的客户,本公司予以指导。
基因表达产物的纯化
通过上述组合筛选出的表达菌株发酵后纯化目标蛋白。由于本系统采用了6His-tag、GST和MBP等亲和配体,可快速纯化出目标蛋白。凡购买本公司试剂盒的客户,本公司予以协助。
四、载体资料
注意:表达载体表达功能区基因测序最终与本公司的DNA测序原始波形图一致(来函备索)。
五、基因高效快速表达试剂盒质量验收标准
一、菌株 E.coli TOP10
E.coli BL21(DE3)plysS
菌株要求:
(1)复苏成活。
(2)无杂菌污染。
(3)抗性与基因型一致。
(4)LB平板菌落为大肠杆菌典型菌落。
(5)转化带氨苄抗性基因质粒时负对照无菌落生长。
(6)转化率大于10的六次方。
(7)8号质粒pTrc-CKS转化E.coli Top 10,IPTG诱导表达量大于30%(可溶)。
(8)9号质粒pET-DsbA转化E.coli BL21(DE3)plysS,IPTG诱导表达量大于30%(可溶)。
二、抗生素
四环素和氯霉素抗性检验可参照《中国药典》进行。
三、表达载体
(1)所有载体均具氨苄抗性。
(2)表达载体大小与说明书一致。
(3)表达载体酶切位点与说明书一致。
(4)表达载体表达功能区基因测序与DNA测序原始波形图一致(来函备索)。
(5)以Maltose binding protein基因为目标基因,所有九种载体均能表达,表达量从5%(分泌型)-40%(胞内可溶)。
(6)质粒无降解,确保转化、酶切、连接等试验成功。
六、相关产品
(1)重组3C protease
包装量:1000 unit/支,其中每1000 unit 3C protease可以切割50mg融合蛋白
(2)基因工程下游蛋白纯化试剂盒
本公司有GST、6His-tag、MBP等亲合层析柱以及阳、阴离子交换、疏水层析和分子筛等蛋白纯化试剂盒。
七、参考文献
Biotechnology (N Y) 1993 Feb;11(2):187-93
A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm.
LaVallie ER, DiBlasio EA, Kovacic S, Grant KL, Schendel PF, McCoy JM.
Genetics Institute, Cambridge, MA 02140.
We have developed a versatile Escherichia coli expression system based on the use of E. coli thioredoxin (trxA) as a gene fusion partner. The broad utility of the system is illustrated by the production of a variety of mammalian cytokines and growth factors as thioredoxin fusion proteins. Although many of these cytokines previously have been produced in E. coli as insoluble aggregates or "inclusion bodies", we show here that as thioredoxin fusions they can be made in soluble forms that are biologically active. In general we find that linkage to thioredoxin dramatically increases the solubility of heterologous proteins synthesized in the E. coli cytoplasm, and that thioredoxin fusion proteins usually accumulate to high levels. Two additional properties of E. coli thioredoxin, its ability to be specifically released from the E. coli cytoplasm by osmotic shock or freeze/thaw treatments and its intrinsic thermal stability, are retained by some fusions and provide convenient purification steps. We also find that the active-site loop of E. coli thioredoxin can be used as a general site for small peptide insertions, allowing for the high level production of soluble peptides in the E. coli cytoplasm.
|
: Appl Environ Microbiol 1998 Dec;64(12):4891-6
|
Expression of active human tissue-type plasminogen activator in Escherichia coli.
Qiu J, Swartz JR, Georgiou G.
Molecular Biology Program, University of Texas, Austin, Texas 78712, USA.
The formation of native disulfide bonds in complex eukaryotic proteins expressed in Escherichia coli is extremely inefficient. Tissue plasminogen activator (tPA) is a very important thrombolytic agent with 17 disulfides, and despite numerous attempts, its expression in an active form in bacteria has not been reported. To achieve the production of active tPA in E. coli, we have investigated the effect of cooverexpressing native (DsbA and DsbC) or heterologous (rat and yeast protein disulfide isomerases) cysteine oxidoreductases in the bacterial periplasm.Coexpression of DsbC, an enzyme which catalyzes disulfide bond isomerization in
the periplasm, was found to dramatically increase the formation of active tPA both in shake flasks and in fermentors. The active protein was purified with an overall yield of 25% by using three affinity steps with, in sequence, lysine-Sepharose, immobilized Erythrina caffra inhibitor, and Zn-Sepharose resins. After purification, approximately 180 microgram of tPA with a specific activity nearly identical to that of the authentic protein can be obtained per liter of culture in a high-cell-density fermentation. Thus, heterologous proteins as complex as tPA may be produced in an active form in bacteria in amounts suitable for structure-function studies. In addition, these results suggest the feasibility of commercial production of extremely complex proteins in E. coli without the need for in vitro refolding.
|
: Protein Sci 2002 Feb;11(2):313-21
|
Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli.
Hammarstrom M, Hellgren N, van Den Berg S, Berglund H, Hard T.
Department of Biotechnology, Royal Institute of Technology (KTH) Center for Physics, Astronomy and Biotechnology, S-106 91 Stockholm, Sweden.
A prerequisite for structural genomics and related projects is to standardize the process of gene overexpression and protein solubility screening to enable automation for higher throughput. We have tested a methodology to rapidly subclone a large number of human genes and screen these for expression and protein solubility in Escherichia coli. The methodology, which can be partly automated, was used to compare the effect of six different N-terminal fusion proteins and an N-terminal 6*His tag. As a realistic test set we selected 32 potentially interesting human proteins with unknown structures and sizes suitable for NMR studies. The genes were transferred from cDNA to expression vectors using subcloning by recombination. The subcloning yield was 100% for 27 (of 32) genes for which a PCR fragment of correct size could be obtained. Of these, 26 genes (96%) could be overexpressed at detectable levels and 23 (85%) are detected in the soluble fraction with at least one fusion tag. We find large differences in the effects of fusion protein or tag on expression and solubility. In short, four of seven fusions perform very well, and much better than the 6*His tag, but individual differences motivate the inclusion of several fusions in expression and solubility screening. We also conclude that our methodology and expression vectors can be used for screening of genes for structural studies, and that it should be possible to obtain a large fraction of all NMR-sized and nonmembrane human proteins as soluble fusion proteins in E. coli.
|
: Protein Expr Purif 1998 Mar;12(2):159-65
|
|
Expression of eukaryotic proteins in soluble form in Escherichia coli.
Zhang Y, Olsen DR, Nguyen KB, Olson PS, Rhodes ET, Mascarenhas D.
Department of Molecular and Cell Biology, Celtrix Pharmaceuticals, Inc., Santa Clara, California 95054, USA.
At the optimum temperature for its growth (37 degrees C), Escherichia coli tends to accumulate heterologous proteins in insoluble form. Fusion protein technology has been used to increase the solubility of overexpressed proteins in this organism, but with variable degrees of success. Fusion to a mutant form of DsbA (DsbAmut) confers higher levels of solubility to heterologous proteins in a reproducible way, even when E. coli is grown at 37 degrees C. We have shown this to be true with a diverse sample of eukaryotic proteins: IGF-I, IGFBP-3, 3C proteinase, TGF beta-2, sTGF beta-RII, BDNF, GDNF, mEGFBP, leptin, and GFP. In addition, we have investigated the effects of charge average and proline content on the solubility of DsbAmut fusions. Coexpression of a protein prolyl isomerase [cyclophilin (L-)] and modification of selected asparagine residues to aspartic acid appear to have beneficial effects on the accumulation of soluble heterologous proteins.
|
: Biotechnology (N Y) 1994 Jun;12(6):601-5
|
Efficient and rapid affinity purification of proteins using recombinant fusion proteases.
Walker PA, Leong LE, Ng PW, Tan SH, Waller S, Murphy D, Porter AG.
Protein Engineering Laboratory, National University of Singapore.
In the affinity purification of recombinant fusion proteins, the rate-limiting step is usually the efficient proteolytic cleavage and removal of the affinity tail and the protease from the purified recombinant protein. We have developed a rapid, convenient and efficient method of affinity purification which can overcome this limitation. In one example of the method, the protease 3C from a picornavirus (3Cpro), which cleaves specific sequences containing a minimum of 6-7 amino acids, has been expressed as a fusion with glutathione S-transferase. The resultant recombinant ‘fusion protease’ cleaves fusion proteins bearing (from the
amino-terminus) the same affinity tail as the fusion protease, a 3Cpro cleavage recognition site, and the recombinant protein of interest. The recombinant protein is purified in a single chromatographic step which removes both the affinity tail and the fusion protease. The advantages over existing methods include much improved specificity of proteolytic cleavage, complete removal of the protease and the affinity tail in one step, and the option of adding any desired amount of fusion protease to ensure efficient cleavage. The potential flexibility of the method is shown by the use of various affinity tails and alternative fusion proteases.